
Economic Operators and the CE Mark on Medical Devices
If you’re a medical device manufacturer, supplier, or importer but aren’t knowledgeable about the European Union Medical Device Regulation (EU-MDR) and In Vitro Diagnostic Regulation (IVDR), it may be high time you did, as their implementation brings about significant changes to the way products get the CE mark (certification), which applies to all economic operators (EOs), spanning device manufacturers, suppliers, and importers that operate throughout Europe.
The Role of EOs
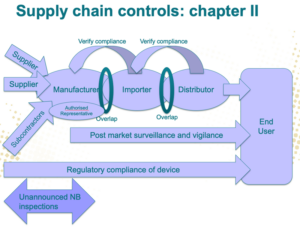
An Economic Operator can be a medical or INV device manufacturer, authorized representative, importer (a natural or legal person that places a device from a third country on the Union market), or distributor (an entity other than the manufacturer or importer that makes a device available on the market) that has specific responsibilities to fulfill. And understanding your position in the supply chain is critical if you want to meet your duties.
Manufacturers and the CE Mark
It is the responsibility of manufacturers to ensure that their products bear the CE mark. Obtaining such certification requires implementing risk analyses and complying with regulations, and proving that they have done so.
Authorized Representatives
Authorized representatives must assess if products need additional safety testing and perform and pass said tests before they are sold to ensure no harm comes to their users.
Responsibilities of the person responsible for regulatory compliance include:
- The conformity of medical devices is checked in accordance with the QM system before delivery.
- Technical documentation is kept up to date.
- Market surveillance is performed in compliance with EU regulations.
- All reporting obligations according to the EU regulations are met.
Importers
Only products that comply with EU regulations and bear the CE mark are allowed to enter the market. As such, importers must ensure that the devices they obtain from manufacturers outside the region have done the required procedures and passed the necessary certifications to ensure compliance.
It Is the first time In a regulation that the Importer Is jointly liable like the manufacturer In placing products In the European Union.
EU national authorities require importers to be well-versed in relevant harmonization acts, enough to obtain written assurance from their respective manufacturers regarding regulatory compliance in the form of signed documentation, such as:
- EU Declaration of Conformity
- Technical documentation
Distributors
Distributors must ensure that only medical devices that conform to all regulations are made available. To do that, they should confirm that all the manufacturers, authorized representatives, and importers implemented the required checks and balances and provided the corresponding documentation.
New Roles and Responsibilities Stipulated in the MDR and the IVDR
All medical devices sold in the region are required to have Unique Device Identification (UDI) numbers under the MDR. Products must be labeled with a production ID per manufacturing batch or series of manufacturing as well. Each device must also have a unique device identifier (DI).
A new database for clinical research, product registration, and postmarket surveillance is also part of the MDR’s new structure. The EUDAMED database will give all concerned companies, users, regulators, and other stakeholders access to the most up-to-date information about all available medical devices in Europe.
Conclusion
As an EO, it is your duty to conduct thorough research on the product you plan to make available to the EU market for regulatory compliance. You must keep all the relevant documentation on hand. And since the medical device industry has undergone several big changes, you need to take time to get familiar with the new procedures and regulations if you wish to be fully compliant.