
The new MDR / IVDR requires specific and new labeling requirements for Medical Devices imported in Europe. We provide a summary of how devices entering the European market should be labelled.
A Comprehensive Guide to EU MDR Labeling Requirements
Medical device manufacturers must provide detailed product labels to ensure patient safety. This article discusses the primary labeling requirements and how new regulations may impact your professional practice.
Purpose of Labeling Requirements
The European Union has mandated that all providers adhere to the Medical Device Regulation (MDR and IVDR).
New labeling requirements means an expansion in standard medical device packaging. Detailed labels and precautionary instructions are needed on top of general labeling requirements. Noncompliance may cause product recalls, fines, and loss of EU market access.
Labeling Roles and Responsibilities
Labels and accompanying documents must be clear, easily comprehensible, contain instructions for use and warnings of potential risks to patients.
The European Commission, in cooperation with each member country’s national competent authorities and accredited conformity assessment bodies, will take point in implementing the MDR.
Labeling Requirement Differences between the MDD and the MDR
The MDR has expanded the list of products under the category of medical devices compared to its predecessor, the Medical Device Directive (MDD).
New regulations cover the following devices:
- Invasive and come into contact with the human body
- Used for aesthetic purposes
- Utilized to administer, readminister, transport, or store medications, bodily fluids, or other substances
- Used in clinical investigations and other products without medical purposes so long as they are listed in Annex XVI (e.g., contact lenses and liposuction and dermatology equipment)
Labeling Symbols Medical Device Manufacturers Must Use
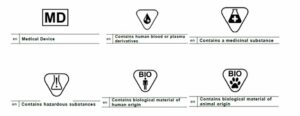
The MDR labeling requirements state that medical device packages should contain symbols that denote its usage and contents, including:
- Medical device symbol indicating the product is for medical purposes, such as disease diagnosis, prevention, monitoring, treatment, or alleviation
- Symbol for products containing biological materials from humans, such as tissues, cells, blood, plasma, or their derivatives
- Symbol for products containing medical or hazardous substances
- Symbol for products containing biological materials from animals
- Symbol stating the products are supplied in a sterile condition, including the specific sterilization method used
- Symbols for translation and repackaging
- Symbol indicating whether the product is intended for single-use, reuse on a single patient, and/or reuse on more than one patient
- UDI symbol, which is not mandatory but must be used when multiple data carriers are present on the medical device label
- Labels of the economic operators of the Authorized Representative and importer
Importer Labeling Requirements
Example of our label:

The importer label must include the name, registered trade name or trademark, registered place of business, and address. The label must be placed in a readable area, on the device (if possible), packaging, or document accompanying the device.
What Is E-Labeling?
E-labeling is the process of using a barcode (QR, 1D, or color) or RFID inlay that points to online information that cannot be displayed on a medical device’s actual label.
The EU has approved the use of electronic IFU (eIFU) since March 2013 for use only with the following device types:
- Devices and accessories for the sole use of professionals
- Software (paper option not required)
- Devices with built-in screen displays (paper option never required)
- Fixed installed medical devices (paper option never required if device has a screen)
- Implantable devices (paper option not required)
Conclusion
While the manner in which medical devices need to be labeled has changed, the stipulations manufacturers must meet are clearly stated in the MDR. In addition, the move will enhance patient protection, which is a boon for all stakeholders, even the device users.