
The CE Mark is mandatory for medical devices Imported to Europe. Learn how your products can get CE marking based on Medical Device Regulations.
When Does Your Medical Device Need CE Marking to Export to Europe?
The European medical technology market was estimated to be worth €140 billion in 2020. This is a promising market for non-EU medical device manufacturers. First, they need a CE Mark according to the risk classification to export to countries in the EU.
What Is CE Marking for Medical Devices?
CE stands for Conformité Européenne. The CE Mark appears on products traded on the extended Single Marker in the European Economic Area. The mark signifies that the product meets the safety, health, and environmental protection requirements set by the European Commission.
The CE marking indicates that the product has met the General Safety and Performance Requirements set by Medical Device Regulations (MDR).
Medical devices are classified into classes, similar to that of the Food and Drug Administration in the U.S. The MDR classes are the following:
Class I
These are considered low-risk devices like bandages, surgical instruments, and wheelchairs.
Class IIa
These are medium-risk devices that require the involvement of a notified body for export. Examples are hearing aids, catheters, and short-term contact lenses.
Class IIb
These products are considered medium to high risk, such as ventilators, long-term contact lenses, and incubators.
Class III
These medical devices are considered high-risk and are subject to strict regulatory requirements. Examples include pacemakers, prosthetics, and other devices that require permanent monitoring.
How Are Medical Devices Classified for CE Marking?
All medical devices undergo stringent technical documentation for CE marking. Factors that affect classification are the characteristics of the devices. Are they non-invasive? Most devices that do not penetrate the body are typically considered Class I. However, some exemptions would result in the product’s higher classification.
Invasive (enter the body through the skin or any orifice) and active (require an energy source) devices are classified higher. Duration of use and reusability also affect classification.
What Is a Notified Body? Do You Need One for CE Marking?
A Notified Body (NB) is a third-party organization designated by an EU country to assess product compliance before it can be sold on the market.
In this case, the NB will assess medical devices and in-vitro diagnostics (IVDs) and their compliance with regulatory requirements.
All medical devices except those that fall under self-certified Class 1 (not sterile, not a measuring device, and not a reusable surgical instrument) require NBs for EU export.
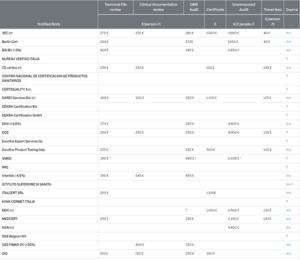
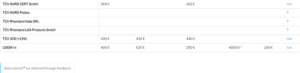
What does a notified body cost?
Qualitiso compared the rates of notified bodies considering days of 8 hours and 2 audit days for a class IIb medical device.
Steps to Obtain CE Marking for Your Medical Device
1. Classify Your Medical Device
The most crucial step is to correctly classify your medical device. The MDR’s decision tree indicates specific rules that will help determine your device’s associated risks and classify it accordingly.
2. Create Quality Management System
A Quality Management System (QMS) includes all policies and procedures related to every step in the design, manufacture, and distribution process of your medical device. This is a mandatory requirement for any company that wishes to enter the medical device market.
3. Create Technical File
Your medical device’s technical file covers all device details to ensure that it meets all MDR requirements. These include:
- Name and address of the company, facilities involved in design and manufacture, and the NB
- Product description and serial number
- EU declaration of conformity
- Instructions for use, parts list, and test results
- Compliance with technical standards and regulations
4. Obtain Certification
Unless the device is under self-certifying class I, you must apply for certification of conformity to essential requirements from an NB.
You can partner with CE marking companies for support throughout these steps.
5. Affix the CE Mark
When all requirements are satisfied, you can create a Declaration of Conformity, and the CE mark will be placed on your medical device.
More Information?
GrowthImports
We provide non-EU medical and in-vitro device manufacturers with independent European-wide importing services compliant with Medical Device Regulation (MDR) and In-Vitro Diagnostic Regulation (IVDR) requirements.
Import your medical goods Into Europe with our hassle-free market access Importing service while maintaining flexibility, compliance and Increased quality standards.
With over 30 years of combined experience, GrowthImports is dedicated to ensure the facilitation of a compliant and smooth international MDR/IVDR compliant importing process in the European market.
GrowthImports BV
Boompjes 5 | 7607 HE Almelo
The Netherlands
T: +31 85 13 08 966
E: info@growthimports.eu
W: www.growthimports.eu